-
INTRODUCCIÓN AL SISTEMA INMUNE HUMANOIntroducción. Conceptos básicos10 Temas
-
Introducción Inmunología
-
Funciones sistema inmune y barreras de defensa
-
Inmunidad innata vs adaptativa
-
Respuesta humoral vs celular
-
Respuesta adaptativa primaria vs secundaria
-
Características de la respuesta inmunitaria adaptativa
-
Concepto de antígeno
-
La respuesta inmunitaria específica es clonal
-
Revisión de los componentes del sistema inmunitario
-
Patología general del sistema inmunitario
-
Introducción Inmunología
-
Células del sistema inmune y diferenciación celular6 Temas
-
Hematopoyesis
-
Las células sanguíneas (I): Granulocitos y Mastocitos
-
Las células sanguíneas (II): Plaquetas, Eritrocitos, Monocitos y Macrófagos
-
Las células sanguíneas (III): Linfocitos (T y B), Células plasmáticas y LGL (Linfocitos NK)
-
Las células sanguíneas (IV): Células dendríticas y proporciones relativas de los tipos celulares
-
Funciones y gestión de receptores para antígenos
-
Hematopoyesis
-
Tejidos del sistema inmune: órganos linfoides 1º y 2º3 Temas
-
Células y mecanismos de la inmunidad innata (I): macrófagos, receptores y mecanismos efectores5 Temas
-
Células y mecanismos de la inmunidad innata (II): linfocitos NK, receptores y mecanismos efectores4 Temas
-
MOLÉCULAS IMPLICADAS EN EL RECONOCIMIENTO DE ANTÍGENOEl receptor de antígeno del linfocito B6 Temas
-
Inmunoglobulinas (I): Formas en la naturaleza, estructura y dominio básico
-
Inmunoglobulinas (II): Clases, formas, variabilidad y tejidos
-
Inmunoglobulinas (III): Enlaces antígeno-anticuerpo y funciones
-
Inmunoglobulinas (IV): Receptores para inmunoglobulinas
-
Inmunoglobulinas (V): Procesamiento de RNA, receptor del linfocito B, complejo co-receptor y activación célula B
-
Inmunoglobulinas (VI): Desarrollo / Diferenciación células B
-
Inmunoglobulinas (I): Formas en la naturaleza, estructura y dominio básico
-
El receptor de antígeno del linfocito T4 Temas
-
Mecanismos de generación de la diversidad de linfocitos T y B9 Temas
-
Genes para las cadenas pesadas y ligeras: Diversidad potencial
-
Reordenamiento de los genes
-
Mecanismos de amplificación de la diversidad
-
Expresión de las inmunoglobulinas en el BCR
-
Maduración de los linfocitos B
-
La cooperación T-B
-
Anomalías/Problemas en el proceso (CLÍNICA)
-
Maduración de los linfocitos B
-
Variabilidad en el repertorio del TcR en linfocitos T
-
Genes para las cadenas pesadas y ligeras: Diversidad potencial
-
El complejo principal de histocompatibilidad (I): estructura proteica, genética y nomenclatura3 Temas
-
El complejo principal de histocompatibilidad (II): Procesamiento y presentación de antígeno, polimorfismo y aplicaciones clínicas5 Temas
-
MOLÉCULAS ACCESORIAS DE LA RESPUESTA INMUNEEl sistema del complemento y sus receptores (I): vía clásica y vía alternativa4 Temas
-
El sistema del complemento y sus receptores (II): vía de las lectinas, vía lítica y regulación3 Temas
-
Moléculas implicadas en la comunicación intercelular (I): citocinas y sus receptores5 Temas
-
Moléculas implicadas en la comunicación intercelular (II): moléculas de adhesión y sus ligandos3 Temas
-
EL SISTEMA INMUNE EN ACCIÓN BLOQUEGeneración de linfocitos T efectores4 Temas
-
Generación de linfocitos B efectores7 Temas
-
Sistema Inmune asociado a mucosas (MALT)9 Temas
-
Introducción MALT
-
Estructura del tejido linfoide asociado a la mucosa intestinal
-
Recirculación y migración selectiva de linfocitos al intestino
-
Barrera epitelial intestinal: Función Inmunológica
-
Receptores para el reconocimiento de patrones moleculares
-
Inmunoglobulinas Poliméricas: IgM + IgA
-
Vías de entrada del antígeno en el intestino
-
Diferenciación de los linfocitos T efectores MALT
-
Tolerancia Oral ante antígenos solubles
-
Introducción MALT
-
La respuesta inmune (I): inmunidad innata e inflamación aguda8 Temas
-
Barreras no específicas frente a infección por microorganismos patógenos
-
Principales leucocitos que paticipan en las inmunidad innata
-
La rección inflamatoria
-
Inflamación aguda
-
Sucesos del proceso inflamatorio
-
Citocinas producidas por macrófagos: efectos locales y sistémicos
-
Efectos de las citocinas en la inmunidad innata
-
Proteínas de fase aguda
-
Barreras no específicas frente a infección por microorganismos patógenos
-
La respuesta inmune (II): mecanismos de la inmunidad específica8 Temas
-
La respuesta inmune (III): respuesta frente a virus, bacterias y hongos, protozoos y helmintos9 Temas
-
Principales enfermedades infecciosas mortales
-
Patógenos infecciosos
-
Respuestas frente a bacterias extracelulares
-
Respuesta frente a bacterias intracelulares
-
Proceso de fagocitosis
-
Respuestas frente a protozoos y helmintos
-
Ciclo vital del plasmodium
-
Respuestas inmunitarias frente a virus
-
Mecanismos de evasión inmunitaria. Virales y bacterianos.
-
Principales enfermedades infecciosas mortales
-
REGULACIÓN e INTRODUCCIÓN A LA INMUNOPATOLOGÍARegulación de la respuesta inmune (I): regulación por moléculas8 Temas
-
Regulación de la respuesta inmune: Introducción
-
Regulación por moléculas: el complemento (I)
-
Proteínas reguladoras del complemento y proceso de regulación (II)
-
Proceso de regulación por complemento (III)
-
Regulación de los anticuerpos preformados
-
Regulación por anticuerpos: Redes idiotípicas
-
Regulación por moléculas de linfocitos T
-
Regulación a nivel bioquímico: ITAM frente a ITIM
-
Regulación de la respuesta inmune: Introducción
-
Regulación de la respuesta inmune (II): regulación por células y sistemas4 Temas
-
El sistema inmune a lo largo del ciclo vital: Inmunosenescencia6 Temas
-
Introducción a la inmunopatología13 Temas
-
Conceptos básicos
-
Inmunodeficiencias
-
Las Inmunodeficiencias (II): Déficits de Complemento
-
Las Inmunodeficiencias (III): Déficits de citocinas en receptores
-
Las Inmunodeficiencias (IV): Déficits de TLR's
-
Las Inmunodeficiencias (V): Déficits de función fagocitaria
-
Las Inmunodeficiencias (VI): Déficit de linfocitos B
-
Las Inmunodeficiencias (VII): Déficits de linfocitos T
-
Inmunodeficiencias primarias (IDP's): Clínica y Diagnóstico
-
Hipersensibilidades
-
Autoinmunidad
-
Trasplantes. Rechazo
-
Inmunidad frente a tumores
-
Conceptos básicos
-
Introducción a la Inmunoterapia8 Temas
-
Vacunas (I): Bases inmunológicas
-
Vacunas (II): Tipos de vacunas
-
Inmunoterapia frente a enfermedades autoinmunes
-
Anticuerpos monoclonales en la terapia del cáncer
-
Otras estrategias anti-tumorales
-
Tratamiento “clásico” de la patología alérgica
-
Inmunoterapia en el trasplante de órganos
-
Tratamiento de inmunodeficiencias primarias
-
Vacunas (I): Bases inmunológicas
Inmunodeficiencias primarias (IDP’s): Clínica y Diagnóstico

La detección clínica de estas patologías, aunque se realiza en la inmensa mayoría en la edad pediátrica, puede tener lugar también en adultos. Existe un “decálogo para la detección de estas patologías” que fue creado por la fundación Jeffrey Model. Es un decálogo de signos clínicos que nos llevan a pensar en una inmunodeficiencia.
Afirmaciones del decálogo:
- Ocho o más “otitis” en un año o menos.
- Dos o más “sinusitis” severas en un año o menos.
- Dos meses (o más) con antibioterapia con poco o ningún efecto.
- Dos o más neumonías en un año.
- Problemas para ganar peso o retraso en el crecimiento (problemas para perder o ganar peso o talla).
- Abscesos recurrentes: dérmicos profundos (o de otros órganos).
- Aftas bucales (o dérmicas) persistentes, en mayores de 1 año.
- Necesidad de antibioterapia intravenosa para superar infecciones.
- Dos o más infecciones de localización profunda: celulitis, meningitis, osteomielitis o sepsis.
- Historia familiar de Inmunodeficiencia primaria.
Con la presencia de uno de estos síntomas ya tienen que saltar las alarmas y es conveniente investigar la posible presencia de enfermedad autoinmune primaria. Si se da más de uno de estos síntomas, ya está claro el diagnóstico de inmunodeficiencia.
Clínica típica general de inmunodeficiencias
- Aumenta el riesgo de infecciones.
- Aumenta el riesgo de tumores: los principales tumores son el linfoma no-Hodgkin y cáncer gástrico porque en algunas inmunodeficiencias no se mueren los linfocitos o hay menos control.
- Aumenta el riesgo de enfermedades autoinmunes (sobre todo en el déficit de IgA).
- Aumenta el riesgo de enfermedades atópicas (sobre todo en el déficit de IgA).
- Frecuentes alteraciones póndero-estaturales.
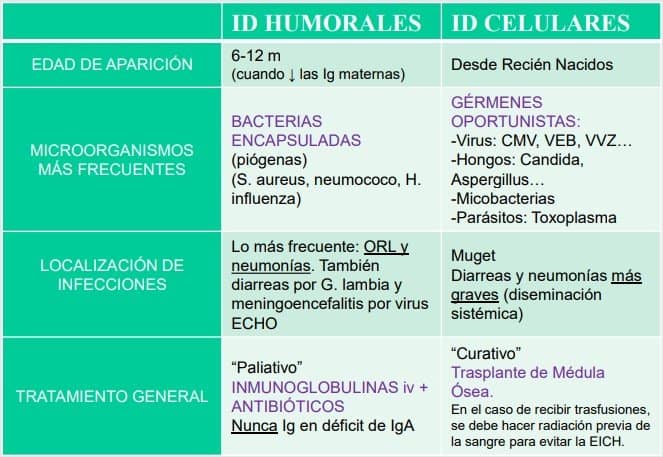
Llega un paciente con un cuadro clínico que nos hace sospechar de una inmunodeficiencia. Miramos la historia familiar. SI no hay casos, puede tratarse de una inmunodeficiencia secundaria.
Puede aparecer un cuadro mixto de inmunodeficiencia con otras enfermedades. Las bacterias o virus que aparecen en un niño con ID nos pueden dar una señal del tipo de ID que padece:
Diagnóstico de IDP’s
- Diagnóstico clínico: historia familiar de inmunodeficiencias, no respuesta a antibióticos, infecciones recurrentes…
- Se realiza un screening básico: pruebas que hacemos en las prácticas, tipo cuantificación de subpoblaciones linfocitarias predominantes y cuantificación de inmunoglobulinas (IgG, IgM e IgA). También se puede realizar un hemograma básico en el que miramos el número de cada grupo linfocitario y hacemos un recuento leucocitario.
- Si los parámetros obtenidos en la prueba son normales, quedan solo unas pocas opciones relacionadas con el complemento y la inmunidad innata. Nos iremos a pruebas más específicas, por lo que lo tendremos que mandar a un sitio de referencia.
- Si es anormal, hay una inmunodeficiencia clara, y solo quedaría determinar si es humoral o celular combinada. Esto tampoco lo hace cualquier médico, se envía a un sitio de referencia.
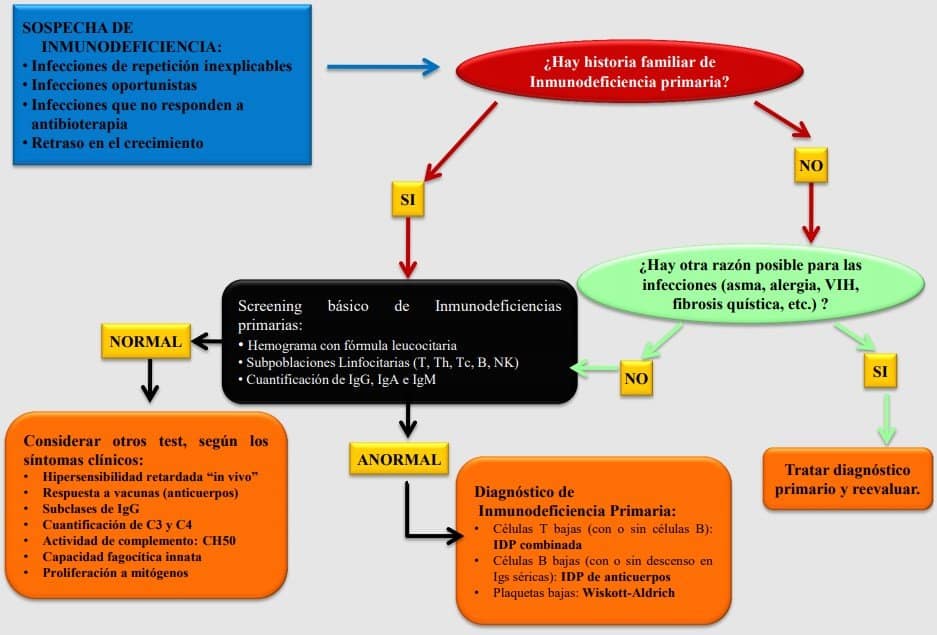
Un paciente llega a consulta con un caso sospechoso de inmunodeficiencia. Primero vemos si hay historia familiar de inmunodeficiencia primaria. Este es un dato clave: si no hay, se descarta la posibilidad de que tenga un a inmunodeficiencia primaria, por lo que sería secundaria. Se buscan posibles causas para los síntomas planteados por nuestro paciente y si no encontramos nada, hacemos un screening básico. Se realiza mediante distintos métodos (subpoblaciones linfocitarias (T, B, NK), cuantificación de inmunoglobulinas, hemogramas…). Si sale todo normal, no podemos descartar todavía que tenga una inmunodeficiencia, y se lleva la muestra a un laboratorio especializado que sea específico para esos parámetros y ver así que es lo que tiene exactamente el paciente.
Ejemplo de IDP: Agammaglobulinemia asociada al X (síndrome de Bruton).
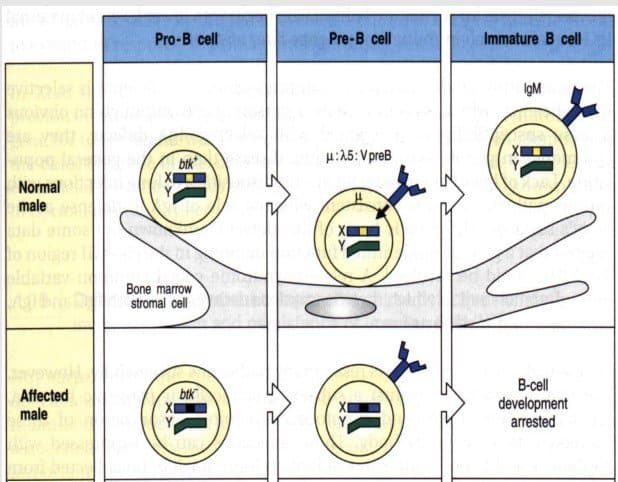
El gen defectuoso es el gen de Btk (tirosin kinasa de linfocitos B). La Btk es necesaria para reordenar la cadena ligera en los linfocitos B en fase pre-B y expresarla en el exterior de la membrana junto a la cadena pesada. (Los linfocitos B maduran en la médula ósea y para pasar del estado de preB a B inmadura, tiene que reordenar la cadena ligera, para lo que es necesaria la Btk).
Los pacientes de síndrome de Bruton (oBTK) no pueden completar la maduración de los linfocitos B, que permanecen en estado pre-B. La célula preB no pasa a B inmadura, por lo que mantiene una cadena ligera complementaria (λ5). No suelen tener linfocitos B en sangre y los que tienen son pocos y en estado pre-B, con una cadena pesada pero sin cadena ligera, por lo que NO producen inmunoglobulinas
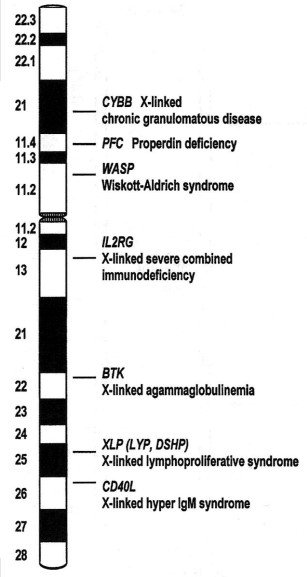
Solo aparece en varones: está ligada al sexo.
Clínica:
- Inicio temprano (en el primer año) de infecciones respiratorias y/o intestinales: en el momento en el que desaparece la IgG materna. A partir del destete, y en caso de lactancia artificial, desde el primer momento de vida.
- Infecciones bacterianas y víricas severas de órganos profundos.
- Edema y artritis (séptica o no) en todas las articulaciones.
- Neutropenia, alopecia y malabsorción (no todos los casos).
Estos niños no tienen inmunoglobulinas ni linfocitos B. Es una enfermedad que debuta muy temprano con infecciones en el tracto respiratorio y gastrointestinal. Suele ocurrir al acabar la lactancia materna o si no hay, antes. Las infecciones son tanto por bacterias como por virus y suele ir acompañada de artritis y de inflamación de las articulaciones (edemas articulares); también presentan retraso en el crecimiento. Hay otros síntomas que no aparecen en todos los casos como la neutropenia.
Laboratorio:
- Niveles séricos de IgG, IgA e IgM muy bajos o indetectables (0).
- Ac naturales: inexistentes.
- Ac vacunales: inexistentes.
- Células B circulantes: <2%, (signo patognomónico, específico de la enfermedad (BTK)).
La prueba de screening o prueba definitiva es la cuantificación de inmunoglobulinas en suero que estarán bajas o totalmente ausentes. Si esto pasa hay que hacer detección linfocitaria por citometría de flujo y comprobar las poblaciones de linfocitos.
Responses